

 Contact Us
Contact Us






 Hospitals
Hospitals
 Doctors
Doctors
 Diagnostic
Diagnostic
 Pharmacy
Pharmacy
 Health Tips
Health Tips
 Blog
Blog
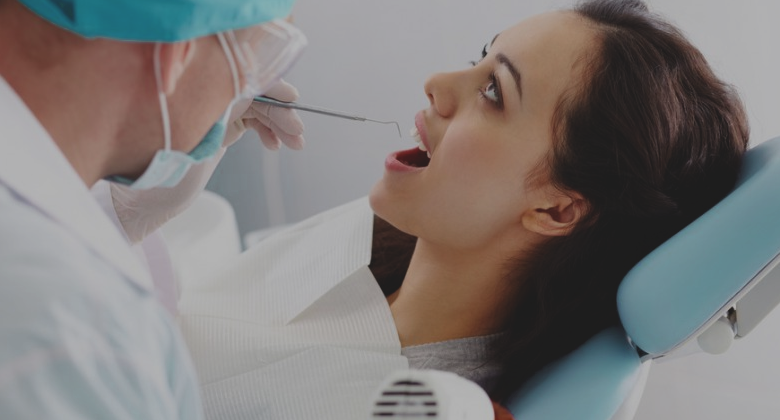
Complete Dental Care Package
From Dental Surgery Clinic
- From: Sunday 15 July 2018
- To: Saturday 15 September 2018
-
If you have any Dental Problem, KayaWell Expert Dr. Nitika Jain is providing free dental consultation for KayaWell users.
FREE
Every First Saturday of Month
#Dental Care



























Comments